
Researchers identify glucose-sensing pathway that connects diabetes and cancer
DATE:2023-07-01
The Warburg effect is a cancer hallmark characterized by excessive glycolysis even when oxygen is present to support mitochondria respiration. This preference is because glycolytic supply of energy and biosynthetic intermediates for other essential bio-macromolecules supports cancer growth. How the Warburg effect is widely achieved amidst tumor heterogeneity is not well understood.
Researchers at the Southern University of Science and Technology (SUSTech) have elucidated a glucose-signaling axis that feeds back to amplify glycometabolism, thereby consolidating the so-called Warburg metabolism to fuel cancer cell growth. These new insights could explain the epidemiological observation that diabetic patients have higher cancer incidence, while suggesting a targetable vulnerability in such patients.

Associate Professor Feng Rao’s research team from the School of Life Sciences at SUSTech recently published a paper that continues the group’s exploration at the frontier of Cullin RING E3 Ligases (CRLs) regulation. In 2020, the Rao lab discovered metabolite-dependent regulation of CRLs by their inhibitor CSN (PNAS, 2020). The same group also found that glucose induces CSN release and CRL4COP1 activation to promote insulin secretion (Nature Communications, 2021).
Their recent work, entitled “Glucose-induced CRL4COP1-p53 axis amplifies glycometabolism to drive tumorigenesis”, has been published in Molecular Cell.
In their paper, the researchers report a glucose-responsive protein modification cascade that reinforces glucose uptake and glycolysis, thereby consolidating the Warburg effect to overcome tumor suppression. Specifically, glucose induces CK2 O-GlcNAcylation, a modification that impedes CK2 phosphorylation of CSN, which in turn is required for CSN to sequester CRL4. Glucose, therefore, elicits CSN-CRL4 dissociation to assemble the CRL4COP1 E3 ligase, which targets p53 for degradation to derepress the glucose transporter GLUT1/4 and many glycolytic enzymes, thereby amplifying glycometabolism through transcriptional reprogramming.
In a mouse tumor model, diet-induced overnutrition accelerates tumorigenesis by promoting CRL4COP1-mediated p53 degradation. These effects of overnutrition are reversed by P28, a peptide inhibitor of COP1-p53 interaction that is under clinical investigation. Therefore, Prof. Rao’s group conclude that glycometabolism can self-amplify via a glucose-induced post-translational modification cascade culminating in p53 depletion. Moreover, such mutation-independent p53 checkpoint bypass may represent the carcinogenic origin and targetable vulnerability of diabetes-associated cancer.
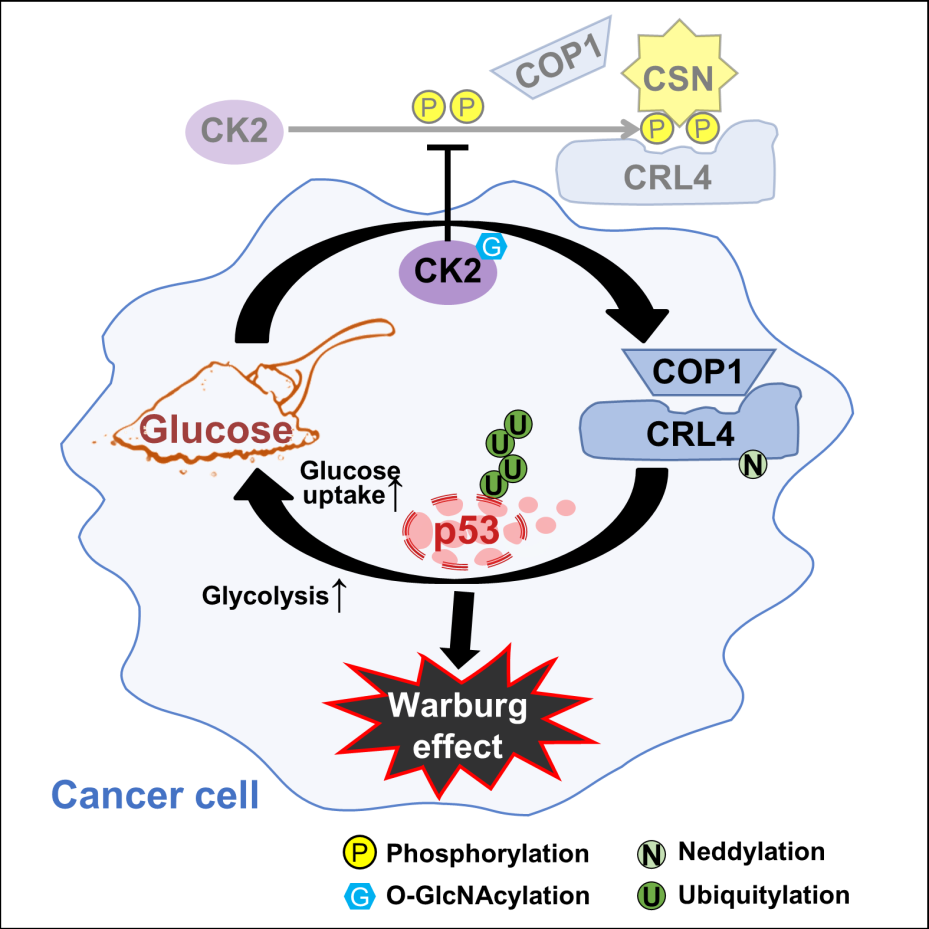
Figure 1. Scheme depicting the molecular mechanisms underlying glucose-induced p53 degradation and its iterative feedback to promote glucose metabolism in cancer. Glucose, via an O-GlcNAcylation-phosphorylation CK2-CSN2 cascade, weakens CSN-sequestration of CRL4 and promotes CRL4COP1 assembly. This leads to p53 degradation and transcriptional up-regulation of glucose uptake and glycolysis. GlcNAc: O-GlcNAcylation; P: phosphorylation; Ub: ubiquitylation; N8: neddylation.
The work was primarily conducted in the laboratory of Assoc. Prof. Feng Rao, in collaboration with Prof. Zhao Li from Tianjin Medical University and Prof. Wang Fengchao from the National Institute of Biological Sciences, Beijing (NIBS). Research Asst. Prof. Su Yang and graduate student Luo Yifan from the Rao lab are the two co-first authors of this paper.
This research was supported by the National Science Foundation of China (NSFC), Shenzhen Municipal Science and Technology Innovation Commission, and the Guangdong Provincial Department of Science & Technology.
Paper link: https://www.cell.com/molecular-cell/fulltext/S1097-2765(23)00432-X
Previous research (PNAS, 2020): https://www.pnas.org/content/117/8/4117.short
Previous research (Nat Comm, 2021): https://www.nature.com/articles/s41467-021-22941-3
Prof. Rao’s webpage: https://faculty.sustech.edu.cn/raof/en/
latest news
-
SUSTech team identifies transposable elements as key determinants of 3D genome structure
Date:2026-03-16
-
SUSTech Researchers Uncover the Molecular Mechanism of C1ql1/BAI3 Assemblies in CF-PC Synapse Development
Date:2025-12-29
-
Researchers discover mechanisms underlying how gravity competes with vision to guide brain’s inner compass
Date:2025-09-22
-
Dynamic changes in transposable elements shape human three-germ-layer differentiation
Date:2025-09-04