
Progress in single-molecule nascent RNA sequencing method reveals landscape of transcription termination in Arabidopsis
DATE:2021-12-08
Transcription termination is the critical final step in RNA synthesis, during which nascent RNA is released from the complex of RNA Polymerase II (Pol II) and DNA template. Termination is essential to prevent uncontrolled readthrough transcription from invading downstream genes.
Decades of extensive research in this area have proposed the allosteric/antiterminator model, the torpedo model, and later a unified model that combines these two to explain the mechanism of polyadenylation signal (PAS)-dependent termination.
Several short-read Illumina-based methods have been developed to analyze nascent RNAs. Nonetheless, their aims focus on the elongating fraction of RNAs before termination rather than the readthrough and cleaved ones. Therefore, these methods cannot distinguish whether the readthrough transcripts are cleaved or not.
Associate Professor Jixian Zhai’s team from the School of Life Sciences at the Southern University of Science and Technology (SUSTech) has revealed the landscape of transcription termination in Arabidopsis using the single-molecule nascent RNA sequencing method.
They observed the delayed termination in atxrn3 mutant and long chimeric transcripts with cryptic splicing in the fpa mutant. Still, loss of CG DNA methylation has no obvious impact on termination in the met1 mutant. Their study, entitled “Landscape of transcription termination in Arabidopsis revealed by single-molecule nascent RNA sequencing,” has been published in the journal Genome Biology.
Identifying the RNA intermediates in transcription termination process
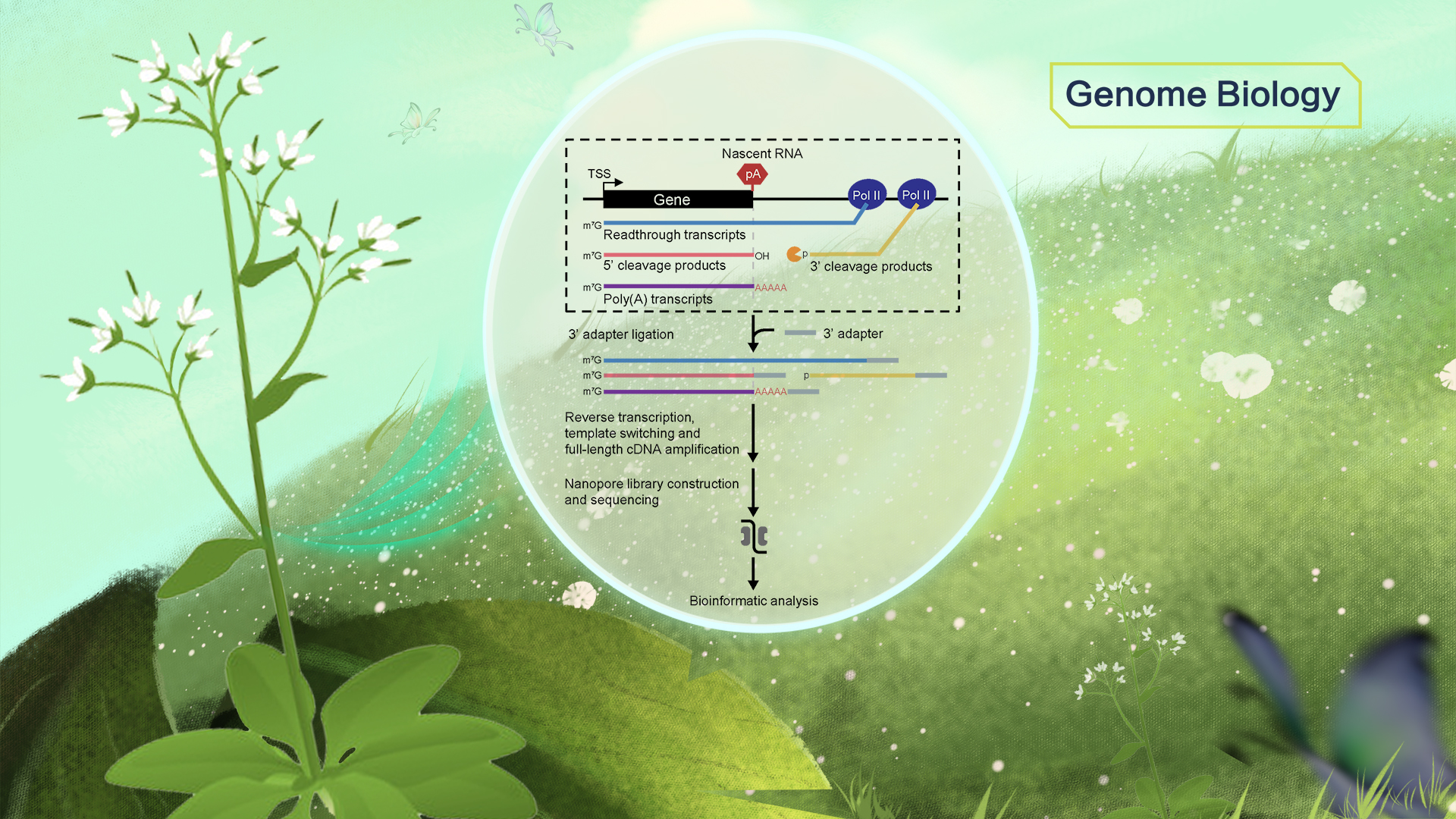
FLEP-seq can distinguish the full-length readthrough, cleaved, and polyadenylated forms of transient RNA intermediates in the termination process (Figure 1). The results show a wide range of termination windows among genes, ranging from ~50 nt to over 1000 nt.
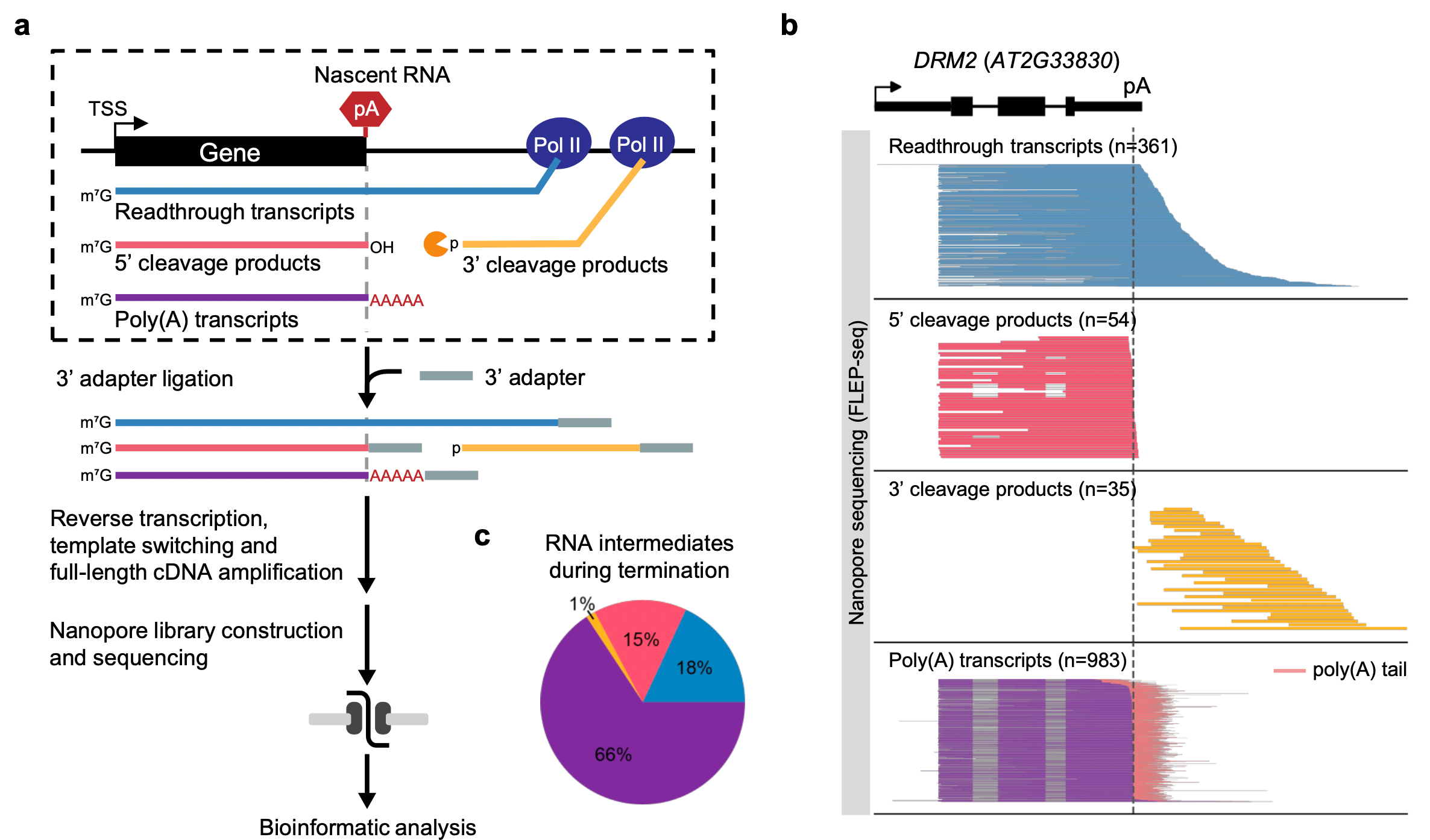
Figure 1. Overview of FLEP-seq that characterizes various forms of transient RNAs during transcription termination at a genome-wide scale.
Transcription termination of Pol II is thought to be a random process that occurs at different distances downstream of the poly(A) site. The distance in which Pol II traveled past the poly(A) site before being released from DNA is described as a “termination window”.
The FLEP-seq data reveal a wide range of termination windows among genes in Arabidopsis, ranging from ~50 nt to over 1000 nt. Prof. Zhai’s team also observed efficient termination before downstream tRNA genes, suggesting that chromatin structure around the promoter region of tRNA genes may block Pol II elongation (Figure 2).

Figure 2. Termination window size distribution in Arabidopsis (left) and downstream tRNA gene effectively promotes transcription termination of upstream genes (right).
The group constructed FLEP-seq libraries for the transcription termination-related genes AtXRN3 and FPA, and the DNA methylation-related gene Met1, respectively. They found that transcription termination was delayed in the atxrn3 mutant and long chimeric transcripts with cryptic splicing in the fpa mutant. However, loss of CG DNA methylation has no noticeable impact on termination in the met1 mutant (Figure 3).
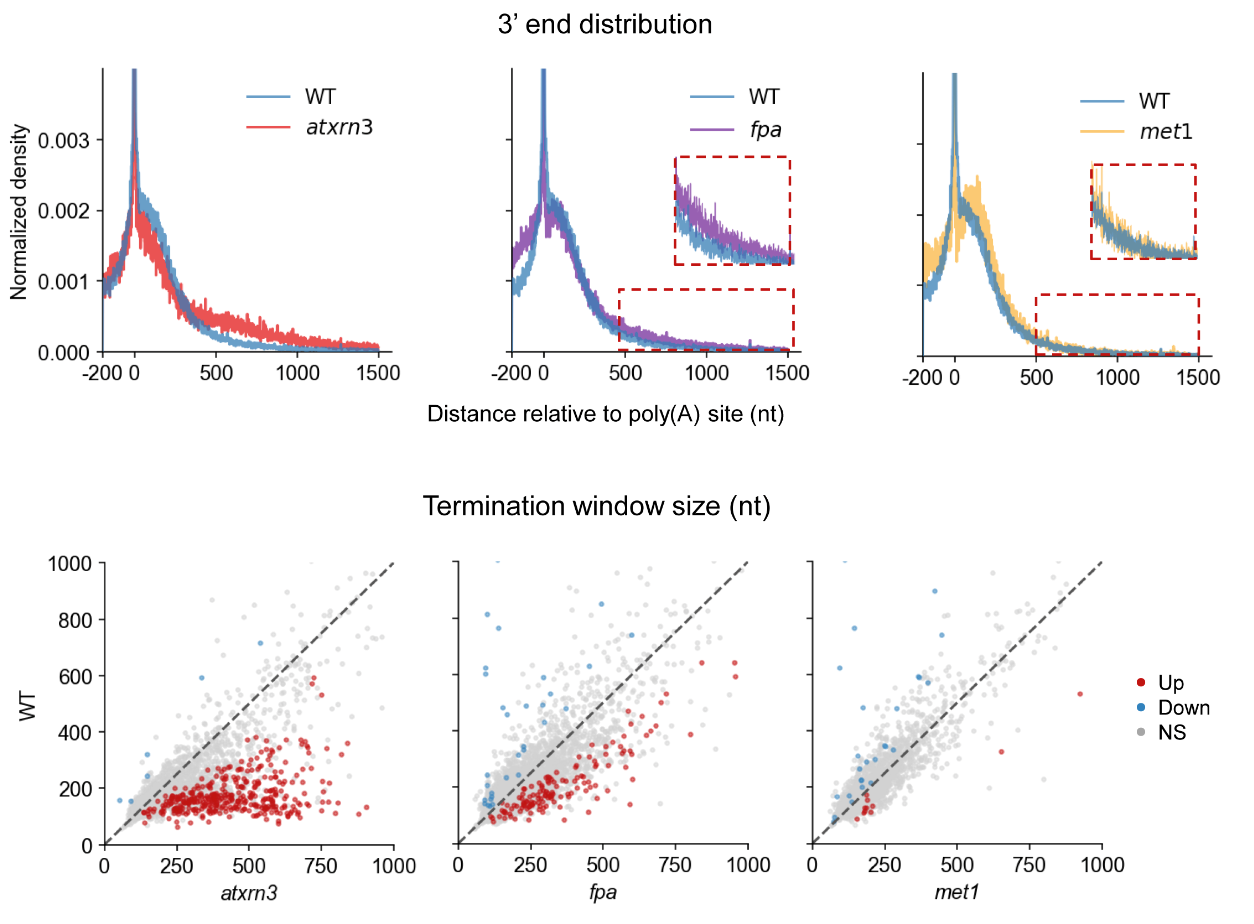
Figure 3. FLEP-seq reveals the transcription termination pattern in the mutant.
Prof. Zhai’s team also observed that readthrough from its gene could continue elongation and pass through the entire downstream gene. It could then be cleaved and polyadenylated, suggesting that readthrough transcription itself could be enough to drive the production of multiple downstream transcripts. This highlights the importance of AtXRN3-mediated transcription termination in the compact Arabidopsis genome (Figure 4).

Figure 4. Example of transcription termination defects in atxrn3 (left) and fpa mutant (right) by FLEP-seq.
Ph.D. students Weipeng Mo and Bo Liu from SUSTech are the co-first authors of this paper. Associate Professor Jixian Zhai from SUSTech is the corresponding author.
Professor Hongwei Guo, Research Associate Professor Yanping Long, and Dr. Jinbu Jia, all faculty members from SUSTech, along with Professor Xiaofeng Cao and Xian Deng from the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences (IGDB, CAS), also made contributions to this work.
This research was supported by the National Key R&D Program of China, the Program for Guangdong Introducing Innovative and Entrepreneurial Teams, the Shenzhen Sci-Tech Fund, the Key Laboratory of Molecular Design for Plant Cell Factory of Guangdong Higher Education Institutes, and the Stable Support Plan Program of Shenzhen Natural Science Fund.
Related links:
Paper link:
Jixian Zhai’s group website:
latest news
-
SUSTech Researchers Uncover the Molecular Mechanism of C1ql1/BAI3 Assemblies in CF-PC Synapse Development
Date:2025-12-29
-
Researchers discover mechanisms underlying how gravity competes with vision to guide brain’s inner compass
Date:2025-09-22
-
Dynamic changes in transposable elements shape human three-germ-layer differentiation
Date:2025-09-04
-
Researchers collaborate to uncover how SOD1 protects lysosome through autophagy
Date:2025-08-26