
SUSTech scholars develops data sketching technique for genomic big data
DATE:2021-04-13
The genomic big data puts a heavy burden on data management and operations. E.g. the size of the NCBI Sequence Read Archive (SRA) database has reached 30 petabytes, which makes conventional sequence analysis tools such as Blast unscalable. Developing more efficient data sketching techniques for genomic big data is urgent. SUSTech scholars have developed a data sketching technique named k-mer substring space decomposition (Kssd) for ‘Omics big data’ analysis.

SUSTech Associate Professor Wenfei Jin led his research group to develop a significantly fast and accurate sketching method. Their research outcomes were published in the high-impact academic journal, Genome Biology, entitled “Kssd: sequence dimensionality reduction by k-mer substring space sampling enables real-time large-scale datasets analysis”.
The proposed technique Kssd can reduce the size of the Omics dataset (including genomics, transcriptomics, metagenomics, etc.) to five orders of magnitudes smaller while still preserving the distance or similarity between two datasets. Therefore, it could be used for real-time analysis (e.g. clustering, quantity control, similar dataset search, etc.) of large-scale Omics datasets. This research also analyzed 1,023,960 bacteria and 1,730 human whole-genome from the NCBI SRA database and identified the mislabeled samples in them.
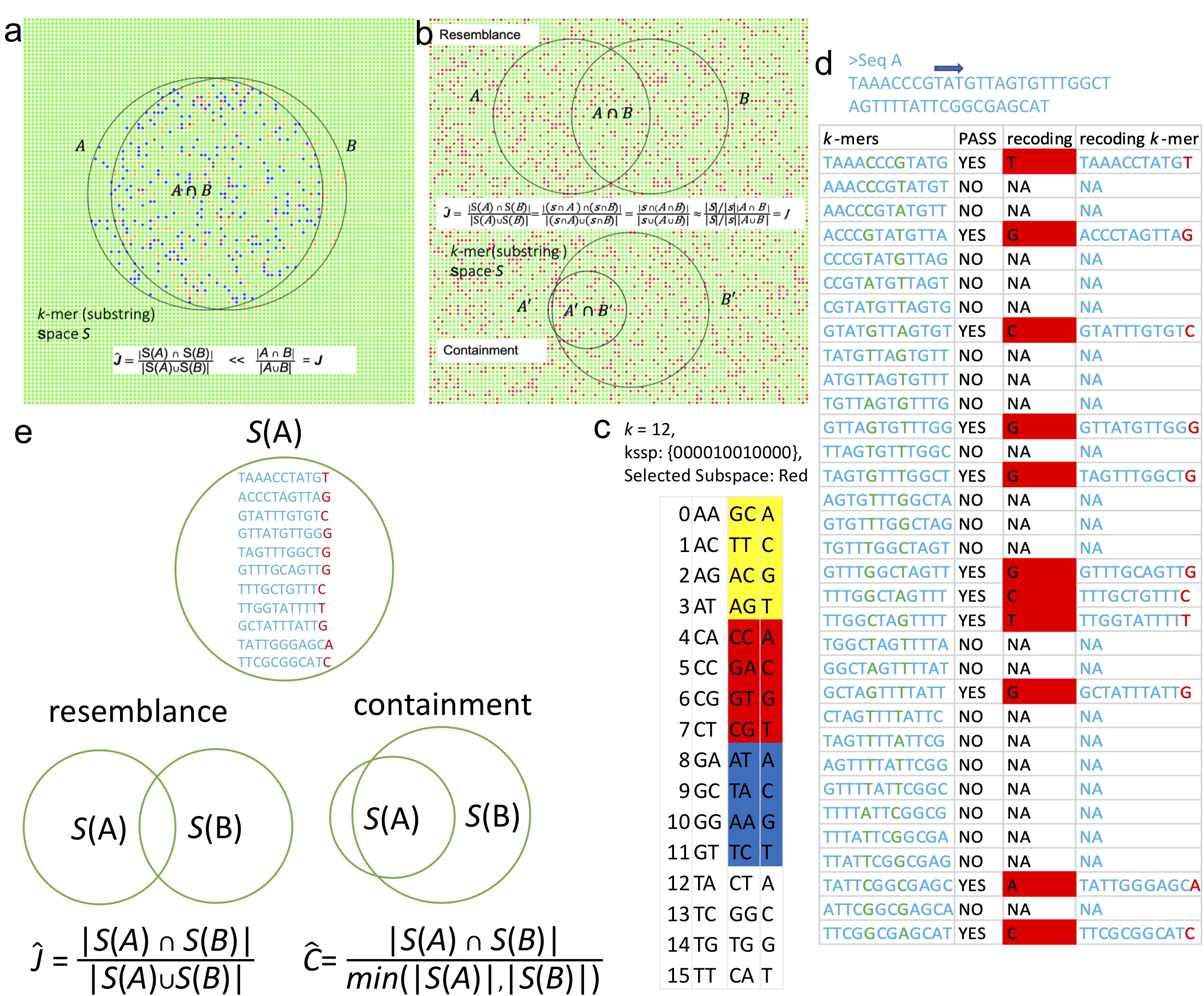
Figure 1. The main idea and algorithm of Kssd
The sequence could be viewed as a set consisting of all the substrings (of a fixed length of k, namely k-mer) contained in this sequence. Sequence dimensionality-reduction is to sample a subset of k-mer from this k-mer set, satisfying that the two sampled subsets of k-mer from the two sequences preserve the similarity or distance of the two sequences. The k-mers subset sampled is termed as sketch and sequence dimensionality-reduction is termed as sketching. The mainstream sketching method “Minhash” obtains sketches by selecting n (typically around 1000) k-mers with minimal hashes from the given sequence. When two sequences are of very different sizes (e.g. a bacteria genome around 5M versus a metagenomics dataset around 5G), the distance and similarity cannot be well preserved by sketch generated using the Minhash approach.
This research proposed the idea of K-mer space sampling. It first draws a subset of K-mer from the K-mer space, and the sketch of a sequence is obtained by intersecting this k-mer subset with the given sequence. Sketches obtained this way preserves the distance and similarity of two given sequences, even when they are of very different sizes, herein greatly extending its application range.
This research further developed the set operations (including intersection, union, and subtraction) between sketches and found that after subtraction of the sketch of the human reference genome, the sketches of human population genomics datasets could be accurately clustered in real-time which was never achieved by other sketching methods before.
The developed technique has broad applications. It can be used in any situation where an approximated similarity measurement is acceptable e.g. phylogenetic tree building, metagenomics containment analysis, and real-time search of large-scale datasets. This technique is a major innovative breakthrough in bioinformatics.
Postdoctoral researcher Huiguang Yi from Wenfei Jin’s group is the first author of this paper, with SUSTech as the first affiliation. Professor Wenfei Jin is the corresponding author. Graduate student Yanling Lin from Wenfei Jin’s group and Professor Chenqi Lin from the Southeast University (SEU) also participated in this research.
This research is supported by the National Key R&D Program of China, Shenzhen Science and Technology Program, China Postdoctoral Science Foundation project, and Jiangsu Planned Projects for Postdoctoral Research Funds.
Article link: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02303-4
latest news
-
SUSTech team identifies transposable elements as key determinants of 3D genome structure
Date:2026-03-16
-
SUSTech Researchers Uncover the Molecular Mechanism of C1ql1/BAI3 Assemblies in CF-PC Synapse Development
Date:2025-12-29
-
Researchers discover mechanisms underlying how gravity competes with vision to guide brain’s inner compass
Date:2025-09-22
-
Dynamic changes in transposable elements shape human three-germ-layer differentiation
Date:2025-09-04